
If you're a scientist transitioning into the biotechnology sector or an entrepreneur developing a novel therapy for an unmet medical need, one term will inevitably surface: Health Technology Assessment (HTA).
While a quick online search may yield basic definitions, these often fail to capture the full scope and real-world implications of HTA in healthcare decision-making. So, what exactly is HTA—and why is it essential for innovators in the life sciences?
The Market Reality
After your product—whether a drug, device, or diagnostic—receives regulatory approval, the next question is: Who’s actually paying for it?
Patients rarely pay directly. Physicians prescribe, but they don’t purchase. The real customers are payers—public and private insurers in the U.S., and government agencies in single-payer systems like the UK, Canada, or Germany. These entities rely on formal HTA processes to ensure that healthcare spending delivers meaningful value.
Historically, many biotech companies assumed that FDA approval would automatically lead to commercial success. But in today’s environment of increasing reimbursement scrutiny, that assumption no longer holds. The recent experience with Alzheimer’s drugs illustrates how payer resistance can derail even FDA-approved therapies (see the example of Aduhelm below).
What Is Health Technology Assessment?
HTA is a structured, evidence-based process used to evaluate whether a healthcare product or service is worth funding. Independent agencies assess:
- Clinical effectiveness
- Economic impact
- Real-world evidence
- Quality of life outcomes
Unlike regulatory bodies such as the FDA—which focus on safety and efficacy—HTA agencies evaluate value for money compared to existing treatments. Their recommendations influence pricing, reimbursement, and market access decisions.
Key HTA Agencies Around the World
- United Kingdom: NICE (National Institute for Health and Care Excellence)
- Canada: CADTH (Canadian Agency for Drugs and Technologies in Health)
- South Korea: HIRA (Health Insurance Review and Assessment Service)
In the United States, there is no national HTA agency. Organizations like ICER (Institute for clinical and economic review)conduct assessments, but their recommendations are not mandatory—leading to greater coverage variation.
Why Do HEOR Professionals Matter?
HTA decisions are grounded in real-world evidence—and this is where Health Economics and Outcomes Research (HEOR) professionals play a pivotal role.
Cost-effectiveness models, real-world data analyses, and quality-of-life studies form the backbone of HTA evaluations. A robust HEOR strategy can be the difference between market access success and failure.
The Bottom Line
HTA enables healthcare systems to make informed, evidence-based decisions about which technologies to fund. For innovators, understanding HTA is not optional—it’s essential. Whether you're developing a breakthrough therapy or planning your market access strategy, aligning with HTA principles is key to navigating today’s value-driven healthcare landscape.
The Rise and Fall of Aduhelm: A Real-World Lesson in Drug Market Access
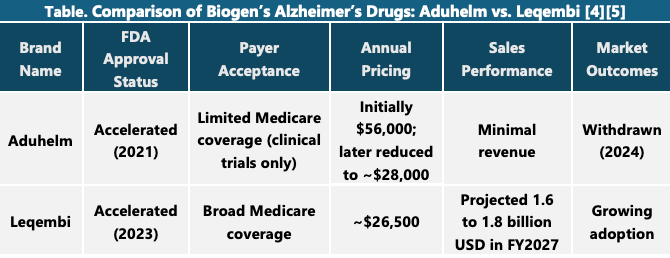
Bioge’s Alzheimer’s drug Aduhelm (aducanumab) represents one of the most controversial episodes in recent pharmaceutical history, highlighting the complex interplay between regulatory approval, payer acceptance, and market viability.
Aduhelm received accelerated approval from the U.S. Food and Drug Administration (FDA) in June 2021, making it the first drug in nearly two decades to target the underlying pathology of Alzheimer’s disease—specifically, the accumulation of amyloid beta plaques in the brain. However, the approval was met with immediate backlash. The FDA had overruled its own independent advisory committee, which had unanimously concluded that there was insufficient evidence of clinical benefit. The fallout included the resignation of several committee members and a congressional investigation that later described the approval process as “rife with irregularities,” citing undocumented communications and inappropriate collaboration between the FDA and Biogen [1].
Despite the regulatory green light, Aduhelm faced significant resistance from payers. The drug launched with a price tag of $56,000 per year, which many experts and advocacy groups criticized as unjustified given the limited and conflicting clinical trial results. The Centers for Medicare & Medicaid Services (CMS) made the unprecedented decision to restrict coverage only to patients enrolled in clinical trials, effectively blocking widespread access. Private insurers followed suit, and many clinicians were hesitant to prescribe the drug due to the uncertainty surrounding its efficacy and the high cost.
As a result, Aduhelm failed commercially, generating minimal revenue despite initial projections that it could become a blockbuster treatment. In early 2024, Biogen announced it would withdraw Aduhelm from the market, ending its post-marketing trials and returning the drug’s rights to its original developer, Neurimmune [2][3].
Biogen has since shifted its focus to Leqembi, a newer Alzheimer’s treatment co-developed with Eisai, which has received full FDA approval and broader payer support. The Aduhelm case is now widely viewed as a cautionary tale—underscoring the importance of robust clinical evidence, transparent regulatory processes, and alignment with payer expectations in the successful commercialization of new therapies [1] [2].
References
[1] Biogen's Controversial Alzheimer's Drug Withdrawn From Market
[3] After disastraous start to launch, Biogen still expects ‘minimal’ sales from Aduhelm this year
[4] Leqembi FDA Approval History
[5] Eisai says out road map to bloackbuster sales projections for Leqembi’s ‘milestone’ 2027 year
Other Posts You Might Like









