Analyzing real-world data (RWD)1 to generate real-world evidence (RWE)2 is an emerging trend in the healthcare community. RWE is routinely used to evaluate a treatment’s real-world effectiveness, safety, and economic value, but it has also been increasingly leveraged in clinical trials. To promote the progression this field, FDA has published multiple draft guidances on RWE since late 2021. The guidance titled “Considerations for the Use of Real-World Data and Real-World Evidence to Support Regulatory Decision-Making for Drug and Biological Products” was recently finalized. It describes FDA’s expectations regarding the utilization of RWD and RWE in non-interventional studies. In this blog post, we’ll explore some of the key takeaways through a series of FAQs.
1. What’s the applicability of this guidance?
This guidance primary focus on non-interventional (observational) studies3 that use only RWD (Figure 1). These studies may be used to help support the approval of a new indication for a drug already approved and other post-approval study requirements. However, FDA also recognizes the utility of RWD in interventional studies4, and specified applicability of other regulations (e.g., investigational new drug application (IND), new drug application (NDA), and biologics license application (BLA)), if RWD was used in interventional studies (Figure 1). Examples include identifying potential participants for a randomized controlled trial, ascertaining endpoints or outcomes in a randomized controlled trial, and serving as a comparator arm in an externally controlled trial5.
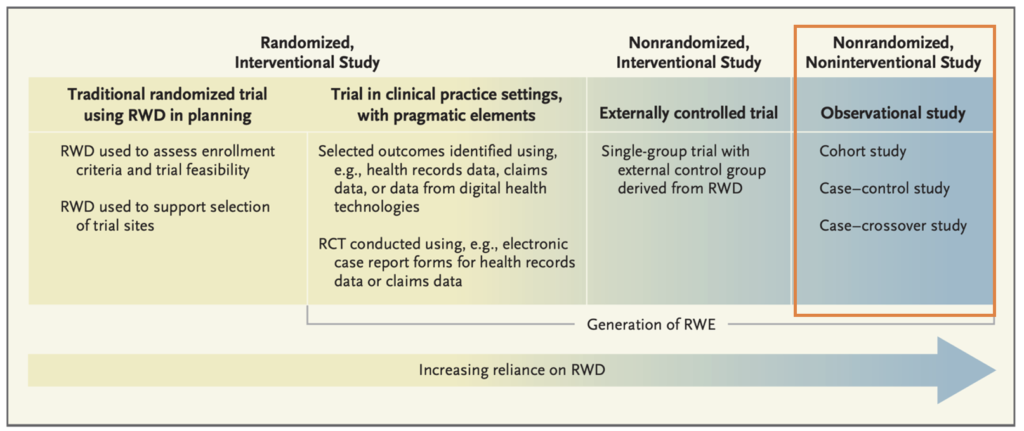
Figure 1. Reliance on RWD in Representative Types of Study Design6. This guidance mainly focuses on non-interventional study. (RCT denotes randomized, controlled trial.)
2. What are FDA’s expectations for non-interventional RWD studies?
In the guidance, FDA outlines high-level regulatory considerations for non-interventional studies using RWD:
- Sponsors should engage with FDA early, which will help address the appropriateness of using a non-interventional study design and the proposed data sources to address the research question of interest.
- Prospective studies may require compliance with regulations on the Protection of Human Subjects and Institutional Review Boards.
- Sponsors should consider and address data privacy issues.
- Transparency regarding data collection and analysis is essential. This includes providing draft protocol and statistical analysis plan (SAP) for FDA review and comment before finalizing them, publishing the protocol on ClincialTrials.gov, justifying the choice of final data sources and analytic approaches, and maintaining audit trails of data and analyses.
- Sponsors should be able submit patient-level data either directly or through third-party data vendors, along with the programming codes and algorithms necessary for FDA to replicate the analyses.
- Rigorously monitoring of the study to ensure the quality and integrity of the data and analyses is required.
- Compliance with post-marketing safety reporting regulations is expected.
3. What other RWE-related guidance FDA has released?
FDA has released several guidances in recent years, as depicted in the timeline diagram below (Figure 2). In addition, FDA initiated the Advancing RWE Program in October 2022 to enhance the quality and acceptability of RWE-based approaches in regulatory decision-making. The program provides sponsors with the opportunity to meet with Agency staff to discuss the use of RWE in medical product development.

Figure 2. FDA Guidances on RWD and RWE
4. What are some successful examples of FDA approvals based on RWE?
On March 23, 2017, avelumab (BAVENCIO®), a programmed death-ligand 1 (PD-L1) blocking human IgG1 lambda monoclonal antibody, received accelerated approval from FDA for the treatment of patients 12 years and older with metastatic Merkel cell carcinoma (MCC), a rare, aggressive skin cancer with a poor prognosis. In the BLA submission, RWD were used as a historical control arm to compare with the single-arm, open label clinical trial7. It marked one of the first instances where a drug was approved by FDA based on supportive effectiveness evidence from RWD. Since then, there have been more than a dozen of successful cases where RWE was submitted in support of FDA approvals of original or supplementary indications, mostly in rare disease and cancer6-12.
Notes and References:
- FDA’s definition: RWD are data relating to patient health status and/or the delivery of health care routinely collected from a variety of sources.
- FDA’s definition: RWE is the clinical evidence about the usage and potential benefits or risks of a medical product derived from analysis of RWD.
- FDA’s definition: Interventional study (also referred to as a clinical trial) is a study in which participants, either healthy volunteers or volunteers with the condition or disease being studied, are assigned to one or more interventions, according to a study protocol, to evaluate the effects of those interventions on subsequent health-related outcomes.
- FDA’s definition: Non-interventional study (also referred to as an observational study) is a type of study in which patients received the marketed drug of interest during routine medical practice and are not assigned to an intervention according to a protocol.
- FDA issued a draft guidance on externally controlled trials using RWE, Considerations for the Design and Conduct of Externally Controlled Trials for Drug and Biological Products.
- Concato, John, and Jacqueline Corrigan-Curay. “Real-world evidence-where are we now?.” The New England journal of medicine 386.18 (2022): 1680-1682.
- Feinberg, Bruce A., et al. “Use of real-world evidence to support FDA approval of oncology drugs.” Value in Health 23.10 (2020): 1358-1365.
- Christina Purpura. “The surge in RWE supported FDA approvals: A look at three FDA decisions.” Aetion.com (2020)
- Arondekar, Bhakti, et al. “Real-world evidence in support of oncology product registration: a systematic review of new drug application and biologics license application approvals from 2015–2020.” Clinical Cancer Research 28.1 (2022): 27-35.
- Maeda, Hideki, and Daniel Bin Ng. “Regulatory approval with real-world data from regulatory science perspective in Japan.” Frontiers in Medicine 9 (2022): 864960.
- Bloomfield‐Clagett, Bartholt, et al. “Use of real‐world evidence in neuroscience‐related new drug and biologics license applications for novel therapeutics.” Clinical Pharmacology & Therapeutics (2023).
- Campbell, Ulka B., Nicholaas Honig, and Nicolle M. Gatto. “SURF: A Screening tool (for sponsors) to evaluate whether Using real‐world data to support an effectiveness claim in an FDA application has Regulatory Feasibility.” Clinical Pharmacology & Therapeutics (2023).



0 Comments